|
2. 1. CONCEPTO.
La podemos definir, según Elliot como la "patología que sigue a una reducción de
la presión ambiente suficiente como para provocar la formación de burbujas a
partir de los gases inertes disueltos en los tejidos".
Es una enfermedad que aparece principalmente en buceadores aunque también puede
afectar a pilotos y trabajadores que lo hagan en ambientes a presión mayor que
la atmosférica como los constructores de pilares de puentes y túneles
submarinos.
2. 2. FISIOPATOLOGIA
El origen de la Enfermedad Descompresiva (E.D.) es la formación de burbujas de
gas inerte, que en el caso del aire comprimido es el nitrógeno (N2) el cual ni
se metaboliza ni combina con ningún sistema biológico y permanece disuelto
aunque inactivo en la sangre y en el organismo durante la descompresión, debido
a un fenómeno de saturación de gas.
Mientras el buceador está en inmersión (fase de compresión), se produce un
incremento de las presiones parciales de los gases inertes en alveolo, sangre,
tejidos y células que van a originar la absorción y disolución (fase de
saturación) de los mismos en el organismo. El N2 es más liposoluble que
hidrosoluble por lo que su difusión es mayor hacia los tejidos ricos en grasa.
La menor tasa de perfusión de estos hace que tarden más tiempo en alcanzar el
estado de saturación obedeciendo a mecanismos de difusión simple.
Según la ley de Henry la cantidad de gas absorbida es proporcional a la presión
parcial del gas. En esta absorción también influyen el tiempo de exposición, el
coeficiente de solubilidad, la temperatura, el riego sanguíneo y la perfusión
tisular.
Clásicamente se pensaba que el CO2 era un factor favorecedor de la aparición de
E.D. pero los estudios desarrollados por Bell reflejan lo contrario, ya que la
hiperventilación debida al CO2 y el efecto vasodilatador del mismo pueden
mejorar la eliminación del gas inerte y con ello disminuir el riesgo de E.D.
Cuando el buceador asciende a la superficie (fase de descompresión) ocurre el
fenómeno inverso: los gases se liberan desde los tejidos al invertirse el
gradiente de presión y salen hacia la sangre y los pulmones (fase de
desaturación).
Por tanto el buceador debe liberar el gas inerte sobrante durante las etapas
finales de la emersión; la cantidad de este gas estará en función de la
profundidad y duración de la inmersión.
Si el proceso de liberación del gas es muy rápido, bien porque se omita la fase
de descompresión, o se haga de forma inadecuada, se pasa de la fase de solución
a la de formación de burbujas (fase de sobresaturación) ya que la
sobresaturación de algunos tejidos puede ser excesiva en comparación con otros
ya desaturados. Cuando la relación entre estos dos conceptos sobrepasa un valor
determinado (razón o cociente de sobresaturación) se alcanza un punto crítico de
sobresaturación ("sobresaturación crítica") a partir del cual el gas cambia de
estado y forma burbujas.
Para que se produzca la E.D.es necesario que la mezcla respirada contenga algún
gas inerte por lo que no puede aparecer E.D. cuando se bucea respirando O2 puro,
pero sí pueden darse casos de E.D. cuando se bucea "a pulmón" (buceo en apnea)
tras inmersiones repetidas.
Para que exista E.D. tienen que producirse burbujas de gas inerte en los tejidos
aunque no está perfectamente determinado el lugar de formación primario ya que
se han observado en las fases iniciales de la descompresión a nivel intersticial
e intralinfático.
Entre las diferentes teorías sobre la fisiopatología de la E.D. la más aceptada
en la actualidad es la que lo explica por la preexistencia de nódulos gaseosos.
Es decir, existirían partículas gaseosas intra o extracelulares absorbidas,
obedeciendo a mecanismos tensoactivos que serían desbordados en caso de agresión
disbárica. Las variaciones de tensión superficial, viscosidad, y densidad, así
como la presencia en disolución de otros gases, actuarían como factores
determinantes de ese equilibrio y serían, en última instancia, los responsables
de que el accidente de buceo determine la E.D.
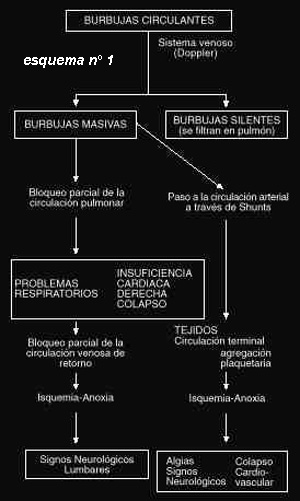
Una vez formadas,"de novo" a partir de núcleos de gas preexistentes o por
alteraciones del volumen crítico, las burbujas circulantes (esquema nº 1)
tienden a eliminarse a través del sistema venoso y los pulmones, ya que la red
alveolocapilar realiza una función de filtro, eliminando por via respiratoria
las burbujas asintomáticas (burbujas silentes).
Las microburbujas pueden permanecer en los tejidos y, dependiendo de su número y
volumen, pueden ser asintomáticas o provocar obstrucción de vasos, disrupción de
tejidos, compresión nerviosa y lesiones cutaneolinfáticas.
Otra posibilidad es el caso de las burbujas extravasculares que emigran a
tejidos vecinos provocando en su trayecto una dislaceración intersticial y con
ello una mayor desnaturalización lipoproteínica lo cual puede perpetuar un
fenómeno de embolismo graso.
Por otra parte, cuando el embolismo venoso es importante, el filtro
alveolocapilar puede ser insuficiente, con lo que se colapsa, provocando un
aumento de la presión en el círculo menor, lo cual dará origen a dos fenómenos:
Por un lado hay una apertura de comunicaciones arteriovenosas, pasando las
burbujas a la circulación arterial y quedando retenidas en los vasos de menor
diámetro, en donde se convierten en burbujas estables o sintomáticas. Estas
afectan principalmente al sistema nervioso central (SNC).
Por otro lado existen burbujas venosas que siguiendo el trayecto de la ázigos
desembocan en los espacios epidurales donde confluyen con otras procedentes del
sistema linfático, y la conjunción de ambas va a provocar un colapso epidural
que se localiza en los segmentos distales de la médula.
Asimismo, la aparición de burbujas en la sangre va a originar fenómenos
reológicos y hemodinámicos que podemos considerar como una auténtica enfermedad
sistémica. La unión de las burbujas con los componentes sanguíneos desencadena
fenómenos electrostáticos de superficie en la interfase "sangre/burbuja"
originando, en primer término, una desnaturalización de las lipoproteinas con
alteración de su estructura, que tendrá como resultado una disminución de la
presión coloidosmótica y subsiguiente pérdida de plasma, con lo que resulta una
hiperviscosidad plasmática, enlentecimiento de la circulación, incremento de la
presión postcapilar, aumento de la permeabilidad precapilar, extravasación
plasmática con hipovolemia y hemoconcentración con mayor hiperviscosidad,
cerrándose el círculo.
Asimismo hay una adhesión plaquetaria además de adherencias de leucocitos y
eritrocitos (efecto "sludge") que obstruyen la circulación capilar y linfática
originando una extravasación plasmática, con lo que incide en el círculo
anteriormente citado incrementándose la hiperviscosidad y la hemoconcentración.
Desde las plaquetas hay una liberación de sustancias vasoactivas principalmente
ADP, PTG, y serotonina que van a favorecer la agregación plaquetaria. Las
proteínas desnaturalizadas activan por la vía intrínseca de la coagulación el
factor Hageman y favorecen la secreción de calicreina y bradicinina que, junto
con la activación concomitante del complemento, van a dar lugar a fenómenos de
quimiotaxis y a la activación del plasminógeno.
También existe una trombocitopenia variable que sirve como parámetro de la
intensidad de la E.D.
Todos estos fenómenos aíslan la burbuja dificultando los intercambios gaseosos
con lo que se impide su resolución.
Las alteraciones bioquímicas "en cascada" de diversos factores de la coagulación
van a dar lugar a un consumo de los mismos que puede llegar a convertirse en un
síndrome de coagulación intravascular diseminada lo que explicaría
fallecimientos tras E.D. masiva (esquema nº 2).
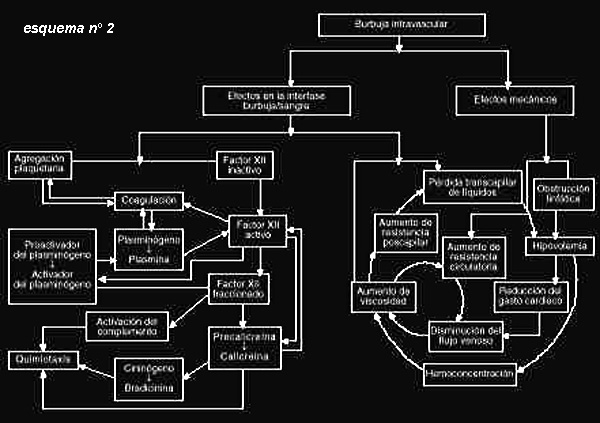
Un método diagnóstico que nos evalúa la gravedad del efecto sobre la coagulación
es el estudio en laboratorio de los D-dímeros plasmáticos.
2. 3. CLINICA
La E.D., en función de su sintomatología ha sido dividida clásicamente en dos
categorías: E.D. tipo I o leve y E.D. tipo II o grave, lo cual se hizo en un
intento de diferenciar los casos y de esta forma poder estandarizar la
identificación, pronóstico y tratamiento.
El Departamento de Medicina Subacuática del Hospital Naval de Bethesda ha
definido la E.D. tipo I como la patología disbárica que se caracteriza por
presentar dolor articular (en inglés "bends"),o rash cutáneo con manifestación
dérmica variada (eritema, exantema, máculas, pápulas, etc), en la que el examen
neurológico es normal y en el que, sometido el paciente a una presión de 2,8 ATA
respirando O2 al 100%, el dolor desaparece dentro de los diez minutos de
recompresión a dicha profundidad.
Sobre la E.D. tipo I no nos vamos a extender debido a la menor importancia de
sus síntomas y por lo tanto, a lo atípico de su aparición en un servicio de
Medicina Intensiva, y solo la citamos con el propósito de que su conocimiento
nos permita valorar su existencia, porque puede ocurrir que una E.D. tipo I
evolucione hacia una E.D. tipo II, lo cual nos debe hacer pensar que estamos
ante un paciente con una E.D. importante.
Las manifestaciones clínicas de la E.D. tipo II implican la posibilidad de
afectación del SNC y periférico (aunque en la mayoría de las series es englobado
en el mismo grupo que las manifestaciones medulares), del sistema
cardiovascular, respiratorio, o gastrointestinal.
Asimismo, una diferenciación que habitualmente no se hace, es entre los síntomas
vestibulares y cerebelosos, a pesar del mejor pronostico de la primera
localización frente a la segunda.
2. 3. 1. Síntomas Neurológicos
Pueden ser debidos a afectación cerebral, cerebelosa, medular, o de los nervios
periféricos. Suele ser mas común entre los buceadores con aire que efectúan
inmersiones de repetición.
Las manifestaciones cerebrales suelen ser más súbitas en su aparición (el 50%
dentro de los 3 primeros minutos) observándose que cuanto mas breve es el
periodo entre la llegada a la superficie y la aparición de los síntomas, mayor
es la severidad del cuadro y peor es el pronóstico.
2. 3. 1. 1. Afectación cerebral
Las manifestaciones clínicas dependen del lugar de la obstrucción vascular y de
la posibilidad de circulación colateral, aunque lo habitual es la afectación de
múltiples puntos localizados principalmente en los lóbulos frontal y parietal.
Cualquier afectación del tejido cerebral va a dar lugar a manifestaciones
análogas a la de cualquier otra patología cerebrovascular, por lo que nos
podemos encontrar con síntomas de hemiplejia, monoplejia, convulsiones focales o
generales, afasia, estados confusionales, cefaleas (por edema cerebral), visión
borrosa o "en túnel", escotomas, disartria, etc.
Como medios diagnósticos, una vez efectuado el tratamiento recompresivo
oportuno, podemos utilizar el EEG, sobre todo en los casos de hemianopsia, y
estudios mediante TAC y RMN que nos pueden localizar las zonas afectadas.
Otras pruebas más sofisticadas, como el empleo de perfusiones con tecnecio, o la
tomografía con emisión de fotones nos pueden ayudar para identificar las
lesiones múltiples y difusas.
2. 3. 1. 2. Afectación cerebelosa
Estas lesiones se pueden manifestar en forma de ataxia, descoordinación, con
típicos signos neurológicos de hipotonía, disminución de los reflejos,
asinergia, dismetría, tremor, diadococinesia y nistagmus.
Los vértigos, habitualmente descritos como vestibulares son probablemente de
origen cerebeloso en la mayoría de los casos.
2. 3. 1. 3. Afectación medular
La localización mas frecuente, y por este orden, corresponde a la zona torácica
media, lumbar superior y cervical baja. Aparece mas comúnmente en pacientes que
presentan síntomas respiratorios ("chokes").
Los signos y síntomas típicos de la afección medular pueden ser precedidos por
un típico "dolor en cinturón" que puede ser el aviso de una enfermedad medular
grave.
La sintomatología más habitual se presenta en forma de paraplejia o paraparesia,
con retención urinaria por parálisis vesical, la cual puede dar origen a un
dolor pélvico que en muchos casos es mal diagnosticado y peor tratado. Asimismo,
los afectados pueden presentar pérdida del control esfinteriano y disestesias en
tronco y abdomen.
En el diagnóstico de la E.D. medular se puede utilizar los potenciales evocados
somatosensoriales para conocer la extensión de la lesión medular además de
servir también para valorar la efectividad del tratamiento.
En relación con la E.D. neurológica se ha observado una mayor prevalencia en los
pacientes que presentan un foramen oval persistente con shunts interauriculares,
que daría lugar a que las burbujas que escapan al filtro pulmonar pasen a la
circulación arterial, aunque trabajos recientes ponen en duda dicha prevalencia.
En los casos en que se sospeche la existencia de un foramen oval persistente
efectuaremos una ecocardiografía transesofágica con contraste para confirmarlo.
2. 3. 1. 4. Afectación de los nervios periféricos
La formación de burbujas en la mielina de los nervios periféricos puede
manifestarse por una desigual afectación motora o sensitiva que afecta
principalmente a los miembros inferiores, siendo la sintomatología mas común las
parestesias, adormecimiento y debilidad motora.
El diagnóstico diferencial entre la afectación neurológica periférica y medular
es importante ya que el pronóstico es mejor en el primer caso.
2. 3. 2. Síntomas vestibulares
La E.D. por localización de la burbuja en oído interno es mas frecuente en el
buceo con mezclas de Helio (principalmente cuando se produce un cambio rápido de
respirar Helio a aire) o Hidrógeno, y mas inusual cuando se bucea con aire.
Pese a ser una rara manifestación de E.D. es importante su conocimiento para
efectuar un diagnóstico diferencial correcto con los accidentes de buceo por
barotraumatismo de oído interno (tabla nº 5), puesto que en el primer caso el
tratamiento recompresivo es fundamental y en cambio la recompresión en el caso
de un barotrauma está totalmente contraindicada ya que ademas de no ser de
utilidad puede agravar el estado del paciente, empeorando el pronóstico desde el
punto de vista funcional del oido interno.
tabla nº 5
|
D. Diferencial de las patologías
del oído interno debidas al buceo |
| |
|
|
Barotrauma (Bt) |
E. D. |
| |
|
| Tipo de buceo: cualquiera
Aparición: ascenso-descenso
Asociación: Bt de oído medio
Gas respirado: aire
Tratamiento: conservador / cirugía
|
Posibilidad de descompresión
En el fondo, ascenso, post buceo
Otros síntomas de E.D.
Principalmente helio o hidrógeno
Recompresión + OHB
|
La E.D. se manifiesta mediante síntomas cocleares, como acúfenos e hipoacusia
neurosensorial, y/o síntomas vestibulares con vértigos, náuseas y vómitos.
Puede suceder que ante un cuadro de E.D. neurológica generalizada, los síntomas
vestibulares pasen desapercibidos o confundidos con síntomas cerebelosos. Para
complementar el diagnóstico diferencial puede ser de utilidad el empleo de la
electronistagmografía, que especificará el origen periférico o central de la
E.D.
2. 3. 3. Síntomas gastrointestinales
Se pueden manifestar por náuseas, vómitos, diarreas o espasmos abdominales. En
los casos mas graves pueden presentarse cuadros de isquemia y hemorragia
intestinal.
Debido a lo infrecuente de su aparición es importante realizar un minucioso
diagnóstico diferencial con otros cuadros gastrointestinales mas frecuentes.
En algunos casos de E.D. fulminante la hemorragia gastrointestinal ha sido la
causa final de la muerte.
2. 3. 4. Síntomas respiratorios y cardiacos
Cuando la liberación de burbujas por parte de los tejidos sucede de forma masiva
puede ocurrir que el lecho vascular pulmonar no sea capaz de evacuar todo el
volumen de burbujas que le llega, dando lugar a manifestaciones pulmonares ("chokes")
con signos y síntomas de distress respiratorio. Para que esto ocurra tiene que
obstruirse un 10% o más del lecho vascular pulmonar.
El gas en el interior de los vasos pulmonares produce el desplazamiento de la
sangre provocando una expansión de los pulmones intravascularmente. El paciente
presenta disnea, taquipnea significativa, dolor subesternal que se agrava con la
inspiración, tos irritativa paroxística y cianosis.
Sin tratamiento, el paciente evoluciona hacia un edema pulmonar (en otras, muy
raras ocasiones, el edema pulmonar ocurre por ahogamiento en los casos de E.D.
severa y de instauración inmediata). Además, el efecto sobre la circulación
pulmonar puede originar una disminución del ritmo cardiaco y de la presión
sanguínea, pudiendo en los casos severos desencadenar un colapso circulatorio y
muerte del paciente.
El diagnóstico clínico será complementado con una radiografía de tórax en el que
se evidencia el edema pulmonar.
En el EKG podemos encontrar una desviación del eje derecho con una elevación de
la onda P, lo cual serán datos a favor de una significativa oclusión de la
circulación pulmonar. La afección cardiaca como sintomatología exclusiva es mas
rara. Se han descrito casos de bloqueo auriculoventricular de primer grado. En
otras ocasiones los cambios en el EKG pueden deberse a burbujas en la
circulación coronaria que van a originar la clínica isquémica correspondiente.
2. 3. 5. Manifestaciones hematológicas
En los casos de una descompresión explosiva podemos encontrar una presencia
masiva de gas en la sangre, lo cual va a originar importantes cambios reológicos
que se van a manifestar con una hemoconcentración grave, llegando a un cuadro de
coagulación intravascular diseminada.
Asimismo aparecen signos y síntomas de shock hipovolémico con hemoconcentración,
hipotensión postural, síncope, mínima (o nula) diuresis, etc..
Los datos del laboratorio mostrarán trombocitopenia, aumento de la VSG,
disminución del sodio y acido láctico, alteraciones enzimáticas, etc...
2. 4. TRATAMIENTO.
2. 4. 1. Evacuación
Al paciente disbárico lo debemos considerar como un "paciente especial" ya que
en su tratamiento se va a requerir el empleo del OHB y por tanto será necesario
disponer de una C.H. Esto es importante, pues no existen en nuestro país
suficientes centros hiperbáricos hospitalarios. Actualmente, los pacientes con
patología disbárica son trasladados a diferentes hospitales generales hasta que
en uno de ellos , y de forma casi siempre casual, alguien recomienda su
evacuación a un centro hiperbárico. Este peregrinaje debiera evitarse, puesto
que el tratamiento fundamental de la E.D. es el recompresivo en C.H. respirando
O2 al 100 % según unas tablas de tratamiento con un protocolo acorde al estado
del paciente.
Antes de comentar el tratamiento en sí haremos algún comentario respecto a la
evacuación del enfermo.
Ante una llamada comunicándonos el envío de un buceador con sospecha de
patología disbárica, debemos recomendar como norma principal que el paciente
respire O2 puro con un flujo de 8-10 L/min (mejor si lo hace en un sistema de
circuito cerrado ya que permite una mayor duración de la botella de O2). Con
esto conseguimos una más rápida eliminación del N2 del organismo así como
contrarrestar el proceso isquémico que la burbuja está provocando.
El paciente debe estar en posición horizontal, bien abrigado y siéndole
suministrado fluidoterapia, principalmente con suero fisiológico y Ringer-lactado.
Si el paciente va a ser trasladado en helicóptero, este no debe sobrepasar los
150 metros como cota máxima de vuelo, y si es por vía terrestre se evitaran
accidentes orográficos mayores de 150 metros de altura. En ambos casos lo que se
pretende evitar es la pérdida de altitud y la consiguiente disminución de la
presión atmosférica, pues ello se traduce en un aumento del diámetro de la
burbuja empeorando el estado, pronóstico y evolución del paciente.
2. 4. 2. Oxigenoterapia Hiperbárica
En cuanto al tratamiento en si, como ya hemos dicho anteriormente, lo principal
y prioritario será la recompresión terapéutica en C.H., donde el paciente
respirará O2 al 100 % según un protocolo de tratamiento con tablas de O2 a baja
presión [N. del A.: Estimo que excede a los fines propuestos en este Tratado el
especificar minuciosamente todos los protocolos de tablas recompresivas de
tratamiento que existen para los accidentes de buceo, ya que dentro de unos
conceptos comunes los hay diferentes en función de la presión,[O2] y duración de
la tabla administrada].
El OHB le llega al paciente bien mediante mascarilla oronasal o a través de una
"capucha" transparente que le cubre totalmente la cabeza, y en los casos mas
graves por intubación endotraqueal. En las C.H. monoplazas, debido a que se
recomprimen con O2 el paciente puede respirarlo directamente.
En todos los casos, la máxima presión a la que se puede respirar O2 al 100 % no
debe exceder de las 3 ATA, lo cual se consigue con el llenado de la C.H. con
aire comprimido, o de O2 en algunas C.H. monoplaza aunque existen también
algunos modelos de estas que se presurizan con aire y el O2 le llega al paciente
mediante los dispositivos anteriormente citados.
Mediante el empleo del OHB no queremos actuar sobre el diámetro de la burbuja,
ya que en la mayoría de los casos y debido al retraso en comenzar el tratamiento
en C.H. no habrá una burbuja de aire sino mas bien un trombo ya organizado y
sobre el cual la acción de la presión no tendrá demasiado efecto para reducir su
tamaño. Más bien la utilización de las tablas sobreoxigenadas se justifica para
contrarrestar la situación de isquemia que la E.D. está provocando. Por tal
motivo en la actualidad se tiende a "saturar" al enfermo con O2, para lo cual lo
mantenemos hasta 8 horas continuadas respirando OHB (aunque haciendo las
oportunas pausas respirando aire para evitar los efectos indeseables
broncopulmonares y neurológicos que puede originar el O2 respirado sin
interrupción durante mas de 2 horas y a una presión de 2,8 ATA).
Asimismo, el OHB tiene una acción reológica mejorando la deformidad
eritrocitaria, lo cual da lugar a una disminución de los fenómenos de "sludge".
Para los casos de E.D. neurológica, principalmente las que afectan a la médula
espinal, que no responden bien al tratamiento con tablas de O2 , últimamente se
están empleando las tablas mixtas O2-helio con óptimos resultados; no obstante,
se precisan más estudios con resultados convincentes que justifiquen el alto
costo que este tipo de tratamiento supone.
Como es norma en Medicina Intensiva, para conseguir los mejores resultados y no
empeorar el pronóstico, evitando asimismo la posibilidad de secuelas, el
tratamiento, en este caso el recompresivo, debe iniciarse tan pronto como sea
posible.
2. 4. 3. Fluidoterapia
Otra actitud terapéutica en la que, como en el punto anterior, todos los autores
están de acuerdo es en el empleo de una fluidoterapia adecuada que solucione la
situación de hipovolemia y trastornos reológicos que tienen lugar, y que
dependerá de la importancia del cuadro.
En los casos leves emplearemos soluciones electrolíticas, principalmente Ringer-lactato,
ya que su osmolaridad está mas próxima a la del plasma que la del suero salino
fisiológico.
En los enfermos más graves utilizaremos expansores del plasma de bajo peso
molecular (dextrano 40.000). En el primer caso no tenemos la necesidad de
corregir urgentemente la hipovolemia, además de que el Ringer lactato posee una
capacidad de expansión volémica débil.
El segundo caso es a la inversa pues ante un paciente grave las soluciones
coloidales artificiales se caracterizan por su poder de expansión volémica
elevada, y de ellas el de poder expansor más rápido es el dextrano 40.000.
Un protocolo puede ser administrar 500 ml inmediatamente y repetirlo a las 4 y
24 horas o bien repetirlo cada 12 horas en las primeras 48 horas. En cualquier
caso no se debe sobrepasar la cantidad de 1,5 gr/Kgr/día, con una diuresis entre
60-100 ml/hora.
Por otra parte el dextrano, ademas de su propiedad expansora, tiene un efecto
anti-sludge, al disminuir la viscosidad sanguínea, principalmente el dextrano
70.000.
En la actualidad tambien se recomienda el empleo de otros coloides artificiales
como el Hidroxietilamidón pero no hay suficiente experiencia para discutir su
idoneidad
Debido al riesgo que existe de sobrecarga vascular, y posible edema pulmonar,
originado por la utilización de los coloides artificiales, es por lo que algunos
autores los desaconsejan además de por la posibilidad de provocar reacciones
anafilácticas.
Finalmente está totalmente desaconsejado el empleo de las soluciones glucosadas
debido a su efecto deletéreo sobre la célula isquémica.
Al contrario de lo que ocurre con respecto al empleo del OHB y de la
fluidoterapia, en donde existe unanimidad de utilización en el tratamiento de la
E.D., a continuación citaremos otras actitudes terapéuticas para las que, o bien
no está suficientemente fundamentado su uso o bien porque al existir estudios
con resultados contradictorios, hace que su indicación tenga mas bien el
carácter de recomendación que de obligación.
2. 4. 4. Corticosteroides
Refiriéndonos a los corticoides, su empleo ha originado gran controversia entre
los distintos autores, además de que los resultados experimentales en modelo
animal son insuficientes para justificar el entusiasmo mostrado en alguna
publicación.
En relación al uso de la metilprednisolona (MP) en forma de "megadosis" (20 mgr/Kg)
hay trabajos que concluyen afirmando la ausencia de interés de la administración
de MP a altas dosis, teniendo en cuenta que la utilización de estos
glucocorticoides es difícil de justificar en razón de sus efectos adversos, en
particular la hipotensión por vasodilatación periférica susceptible de
comprometer la perfusión medular, además de poder aumentar la sensibilidad del
paciente a la neurotoxicidad del OHB (efecto "Paul Bert").
Con respecto a la dexametasona, recientemente se ha comprobado la ineficacia de
su empleo "en bolo" en el tratamiento del edema cerebral originado en la E.D.
Por otra parte y para conseguir un inmediato efecto antiinflamatorio sí se
recomienda que en la fase pre-hospitalaria utilicemos hemisuccinato de
hidrocortisona en dosis de 1 gr por via iv ,ya que su pico sérico es alcanzado
rápidamente (el nivel de máxima concentración sérica se obtiene en 1 hora).
2. 4. 5. Antiagregantes plaquetarios
En lo que respecta a la corrección de los trastornos de la coagulación, la
indicación de los antiagregantes plaquetarios (AAP) es siempre controvertida en
razón de la ausencia de resultados de ensayos clínicos controlados que puedan
legitimar su prescripción. En todo caso se pueden utilizar mas como medida
preventiva que por su actividad terapéutica.
De entre todos los AAP solo dos han demostrado su actividad: El acido acetil
salicílico (AAS) y la ticlopidina. Por tal motivo en el supuesto de utilizar
algún AAP emplearemos el AAS a dosis bajas (150-300 mgr) y debiéramos
adminístralo en el momento de la aparición de los primeros síntomas de E.D. para
evitar la posibilidad de trombosis intravasculares.
2. 4. 6. Heparina
Otro fármaco utilizado en la corrección de los posibles trastornos de la
coagulación es la heparina, de la que algunos autores proponen su utilización
principalmente en los casos de E.D. neurológica, aunque para otros la falta de
ensayos controlados, así como sus resultados inciertos y todo ello unido a la
posibilidad de que podamos agravar fenómenos hemorrágicos, facilitados por las
burbujas de aire o de origen laberíntico, hace que su uso esté localizado en
determinados centros hiperbáricos.
La heparina nunca será utilizada en la fase pre-hospitalaria, sino algunas horas
después del ingreso incluso, tras el tratamiento recompresivo. La escuela
francesa recomienda el empleo de heparina subcutánea a dosis de 0,2-0,3 mL
aunque las mismas estarán en función de las pruebas de coagulación.
2. 4. 7. Otras Actitudes Terapéuticas
Con respecto a los analgésicos, están totalmente prohibidos, pues podrían
enmascarar un dolor muscular que fuera un síntoma-guía que facilitase el
diagnóstico.
Sí puede ser útil la administración de vitamina-E para prevenir la
neurotoxicidad del OHB en pacientes tratados con glucocorticoides.
Por último, en los casos de cuadros neurológicos con parálisis vesical es
necesario su sondaje pues no siempre hay recuperación en el primer tratamiento
recompresivo, lo que hace preciso dicho sondaje vesical aunque retrase la
normalización de la función esfinteriana.
|
Tabla nº 6: Tratamiento de la
E.D. |
| |
|
| Básico: |
|
| |
* Recompresión en cámara hiperbárica.
* Oxigenoterapia hiperbárica.
* Fluidoterapia: Sol salina, Ringer-lactato, Dextranos.
* Postural: Horizontal. |
| Coadyuvante: |
|
| |
* Antiagregantes plaquetarios: AAS,
ticlopidina.
* Hemisuccinato de hidrocortisona.
* Sondaje vesical.
* Vitamina-E. |
| Discrecional: |
|
| |
* Corticoides: Metil-prednisolona,
dexametasona
* Heparina. |
2.4.8. Tratamiento Post-Intensivo
Una vez superado el cuadro agudo, el paciente, en función de su estado o de las
posibles secuelas que presente - ya que un porcentaje notable de casos con
afectación medular o del SNC no consiguen una satisfactoria mejoría después del
tratamiento inicial - puede necesitar continuar con OHB siguiendo un protocolo
de 10 sesiones, a 2-2,5 ATA de presión y con una duración de 90 min/sesión, así
como tratamiento rehabilitador.
Se recomienda un seguimiento de al menos 2 años para valorar la eficacia del
tratamiento administrado.
|